Ein neues 'Periodensystem' für Nanomaterialien
Neue Simulation für Wissenschaftler entscheidet, welche Moleküle am besten miteinander interagieren
Der Ansatz wurde von Daniel Packwood vom Institute for Integrated Cell-Material Sciences (iCeMS) der Universität Kyoto und Taro Hitosugi vom Tokyo Institute of Technology entwickelt. Dabei werden die chemischen Eigenschaften von Molekülen mit den durch ihre Wechselwirkung entstehenden Nanostrukturen verbunden. Eine maschinelle Lerntechnik erzeugt Daten, die dann zur Entwicklung eines Diagramms verwendet werden, das verschiedene Moleküle nach ihren nanogroßen Formen kategorisiert. Dieser Ansatz könnte Materialwissenschaftlern helfen, die geeigneten Moleküle für die Synthese von Zielnanomaterialien zu identifizieren.
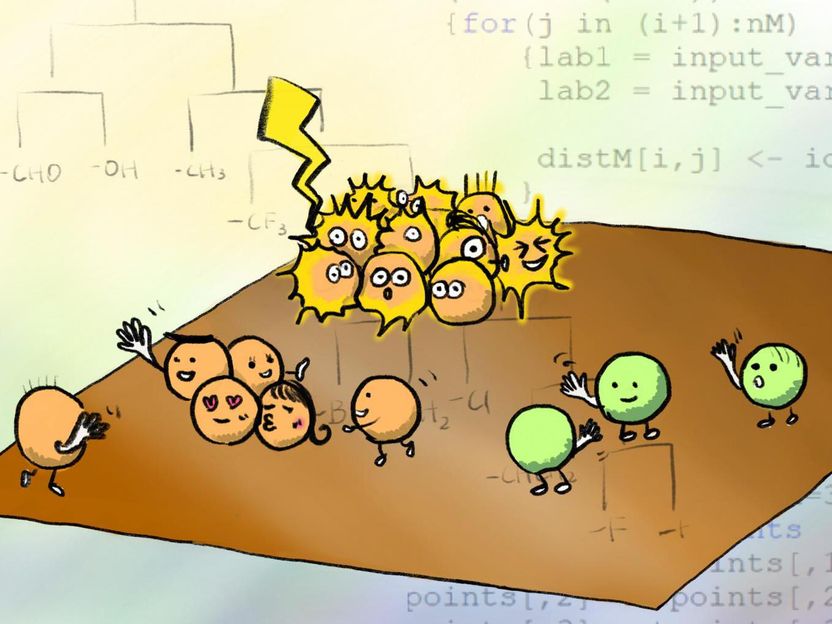
Moleküle interagieren und richten sich bei der Selbstassemblierung aneinander aus. Diese neue Simulation ermöglicht es, herauszufinden, welche Moleküle am besten miteinander interagieren, um Nanomaterialien herzustellen, wie z.B. Materialien, die als elektrischer Nanodraht funktionieren.
Illustration by Izumi Mindy Takamiya
Die Herstellung von Nanomaterialien mit einem Bottom-up-Ansatz erfordert das Finden von "Vorläufermolekülen", die bei der Selbstassemblierung miteinander interagieren und sich korrekt ausrichten. Aber es war eine große Herausforderung zu wissen, wie Vorläufermoleküle interagieren und welche Formen sie bilden werden.
Die Bottom-up-Fertigung von Graphen-Nanobändern wird aufgrund ihrer Einsatzmöglichkeiten in der Elektronik, im Tissue Engineering, im Bauwesen und in der Bio-Imaging-Branche stark nachgefragt. Eine Möglichkeit, sie zu synthetisieren, ist die Verwendung von Bianthracen-Vorläufermolekülen, an die Brom gebunden ist. Die Bromgruppen interagieren mit einem Kupfersubstrat zu nanogroßen Ketten. Wenn diese Ketten erhitzt werden, verwandeln sie sich in Graphen-Nanobänder.
Packwood und Hitosugi testeten ihren Simulator mit dieser Methode zur Herstellung von Graphen-Nanobändern.
In das Modell wurden Daten über die chemischen Eigenschaften einer Vielzahl von Molekülen eingegeben, die an Bianthracen gebunden werden können, um es zu "funktionalisieren" und seine Interaktion mit Kupfer zu erleichtern. Die Daten durchliefen eine Reihe von Prozessen, die schließlich zur Bildung eines "Dendrogramms" führten.
Dabei zeigte sich, dass die Bindung von Wasserstoffmolekülen an Bianthracen zur Entwicklung starker eindimensionaler Nanoketten führte. Fluor-, Brom-, Chlor-, Amidogen- und Vinylgruppen führten zur Bildung mäßig starker Nanoketten. Trifluormethyl- und Methylgruppen führten zur Bildung von schwachen eindimensionalen Molekülinseln, Hydroxid- und Aldehydgruppen zur Bildung von starken zweidimensionalen kachelförmigen Inseln.
Die im Dendrogramm erzeugten Informationen änderten sich aufgrund der zur Verfügung gestellten Temperaturdaten. Die oben genannten Kategorien gelten, wenn die Wechselwirkungen bei -73°C durchgeführt werden. Die Ergebnisse änderten sich bei wärmeren Temperaturen. Die Forscher empfehlen, die Daten bei niedrigen Temperaturen anzuwenden, wo der Einfluss der chemischen Eigenschaften der funktionellen Gruppen auf die Nanoformen am deutlichsten ist.
Die Technik kann auf andere Substrate und Vorläufermoleküle angewendet werden. Die Forscher beschreiben ihre Methode als analog zum Periodensystem der chemischen Elemente, das die Atome nach ihrer Bindung gruppiert. "Um jedoch wirklich zu beweisen, dass die Dendrogramme oder andere Informatik-basierte Ansätze für die Materialwissenschaft so wertvoll sein können wie das Periodensystem, müssen wir sie in ein echtes Bottom-up-Nanomaterial-Fertigungsexperiment einbeziehen", schließen die Forscher in ihrer Studie. "Wir verfolgen diese Richtung derzeit in unseren Labors."
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft



























































