Katalysatorforschung: Molekulare Sonden erfordern hochgenaue Rechnungen
Forscher setzen fortgeschrittene Methoden mit Hybridfunktionalen zur Analyse der aktiven Zentren ein
Katalysatoren machen viele Technologien überhaupt erst möglich. Um heterogene Katalysatoren weiter zu verbessern, bedarf es der Analyse der komplexen Prozesse an ihrer Oberfläche, wo sich die aktiven Zentren befinden. Forschende des Karlsruher Instituts für Technologie (KIT) haben mit Kollegen aus Spanien und Argentinien dabei einen entscheidenden Fortschritt erzielt: Wie sie nun in der Zeitschrift Physical Review Letters berichten, setzen sie Rechenmethoden mit sogenannten Hybridfunktionalen ein, die eine zuverlässige Interpretation experimenteller Daten ermöglichen.
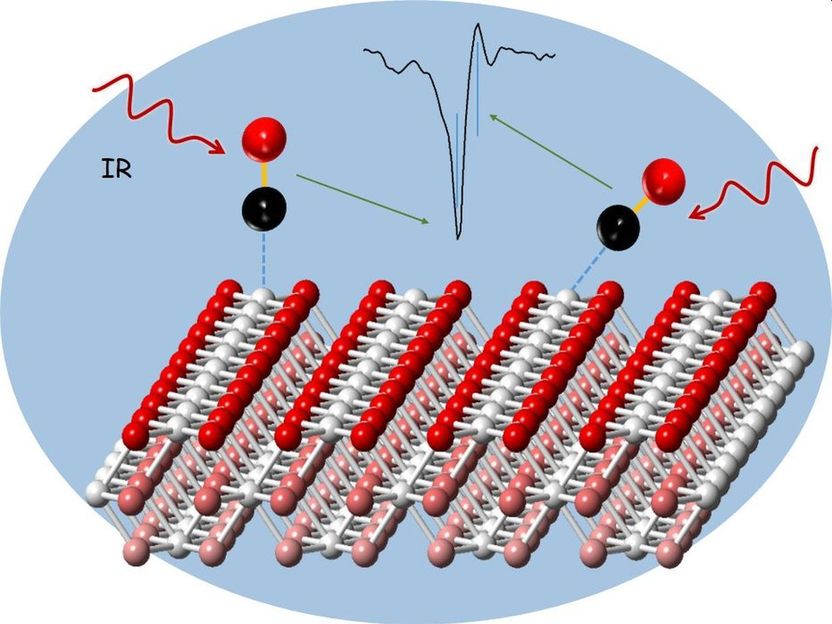
Analyse eines Ceroxid-Katalysators mit Kohlenmonoxid-Sondenmolekülen und Infrarot-Reflexions-Absorptions-Spektroskopie.
IFG/KIT
Viele wichtige Technologien, beispielsweise zur Energieumwandlung, zur Emissionsreduktion oder zur Produktion von Chemikalien, funktionieren nur mit passenden Katalysatoren. Daher gewinnen hocheffiziente Materialien für die heterogene Katalyse immer mehr an Bedeutung. Bei der heterogenen Katalyse liegen der als Katalysator fungierende Stoff und die miteinander reagierenden Stoffe in verschiedenen Phasen vor, beispielsweise fest und gasförmig. Die Materialzusammensetzungen lassen sich mithilfe verschiedener Methoden bereits zuverlässig bestimmen – die Prozesse, die direkt an der Katalysatoroberfläche ablaufen, entziehen sich hingegen noch den meisten Analyseverfahren. „Aber gerade die hochkomplexen chemischen Prozesse an der alleräußersten Haut der Katalysatoren sind entscheidend“, erklärt Professor Christof Wöll, Leiter des Instituts für Funktionelle Grenzflächen (IFG) am KIT, „denn dort befinden sich die aktiven Zentren, an denen die katalysierte Reaktion abläuft.“
Genaue Untersuchung der Oberfläche von Pulverkatalysatoren
Zu den wichtigsten heterogenen Katalysatoren gehören Ceroxide, das sind Verbindungen des Seltenerdmetalls Cer mit Sauerstoff. Sie liegen als Pulver vor, bestehend aus Nanopartikeln mit kontrollierter Struktur. Die Form der Nanopartikel beeinflusst die Reaktivität des Katalysators wesentlich. Um die Vorgänge an der Oberfläche solcher Pulverkatalysatoren zu untersuchen, verwenden Forschende seit einiger Zeit Sondenmoleküle, beispielsweise Kohlenmonoxidmoleküle, die sich an den Nanopartikeln anlagern, und messen die Proben mit Infrarot-Reflexions-Absorptions-Spektroskopie (IRRAS). Die Infrarotstrahlung regt Molekülschwingungen an. Anhand der Schwingungsfrequenzen der Sondenmoleküle lassen sich detaillierte Informationen über die Art und Zusammensetzung der katalytischen Zentren gewinnen. Die Interpretation der experimentellen IRRAS-Daten war bisher allerdings problematisch. Denn gerade für die technologisch relevanten Pulverkatalysatoren sind viele Schwingungsbanden zu beobachten, deren genaue Zuordnung schwierig ist. Theoretische Berechnungen waren dabei bisher nicht wirklich hilfreich, weil die Abweichung vom Experiment – auch im Fall von Modellsystemen – so groß war, dass sich die experimentell beobachteten Schwingungsbanden nicht zuverlässig zuordnen ließen.
Lange Rechenzeit – hohe Genauigkeit
Forschende am Institut für Funktionelle Grenzflächen (IFG) und am Institut für Katalyseforschung und -technologie (IKFT) des KIT haben nun in einer internationalen Kooperation mit Kolleginnen und Kollegen aus Spanien und Argentinien, koordiniert von Dr. M. Verónica Ganduglia-Pirovano vom Consejo Superior de Investigaciones Científicas (CSIC) in Madrid, ein wichtiges Problem der theoretischen Analyse identifiziert und gelöst. Wie die Wissenschaftlerinnen und Wissenschaftler in der Zeitschrift Physical Review Letters berichten, haben sie anhand systematischer theoretischer Studien und der Validierung der Ergebnisse an Modellsystemen gezeigt, dass die bisher eingesetzten theoretischen Methoden grundlegende Schwächen aufweisen. Solche Schwächen sind generell zu beobachten bei Rechnungen mit der Dichtefunktionaltheorie (DFT), einer Methode, mit der sich, beruhend auf der Dichte der Elektronen, der quantenmechanische Grundzustand eines Vielelektronensystems bestimmen lässt. Wie die Forschungsgruppe feststellte, lassen sich die Schwächen mit sogenannten Hybridfunktionalen überwinden, die DFT und Hartree-Fock-Methode, ein Näherungsverfahren in der Quantenchemie, miteinander kombinieren. Die Rechnungen werden dadurch zwar ziemlich aufwendig, aber auch hochgenau. „Zwar sind die für diese neuen Methoden erforderlichen Rechenzeiten um etwa einen Faktor 100 größer als die für konventionelle Verfahren“, erläutert Wöll. „Aber dieser Nachteil wird durch die hervorragende Übereinstimmung mit den experimentellen Systemen mehr als ausgeglichen.“ Am Beispiel nanoskaliger Ceroxid-Katalysatoren demonstrierten die Forschenden diesen Fortschritt, der wesentlich dazu beitragen kann, heterogene Katalysatoren wirksamer und langlebiger zu machen.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft



























































