Schmierstoffe am Computer charakterisieren und designen
Berechnungsmodelle auf Basis von Molekül-Struktureigenschaften
Geschmierte Wellen, Lager oder Getriebe laufen nur dann »wie am Schnürchen«, wenn die Bauteile auf einem perfekten Schmierfilm gleiten und dabei möglichst wenig Reibung, Verschleiß und Energieverlust erzeugen. Dafür müssen die Ingenieure das Verhalten des Schmierfilms im sogenannten Tribokontakt kennen, was mit Experimenten kaum zu messen ist. Das Fraunhofer IWM, MikroTribologie Centrum µTC, macht darum Schmierstoffeigenschaften mittels atomistischer Methoden berechenbar und hat jüngst spannende Erkenntnisse zu einer der zentralen Kenngrößen, der Druckabhängigkeit der Schmierstoffviskosität, in der wissenschaftlichen Fachzeitschrift Physical Review Letters veröffentlicht.
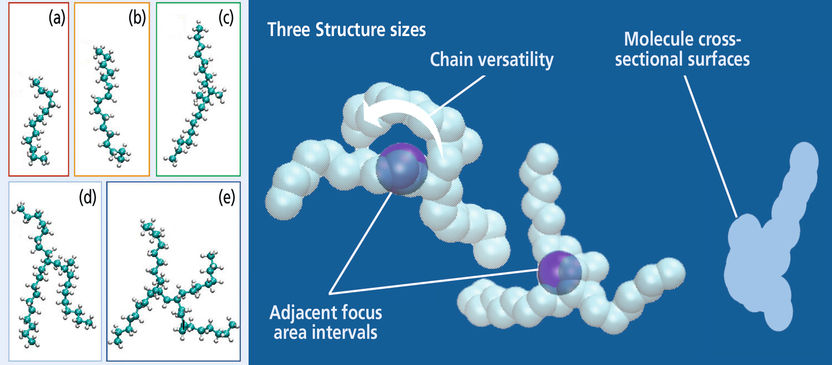
Alkan-Moleküle, deren Viskosität mittels Molekulardynamiksimulation bei Hochdruck- und Hochtemperatur-Bedingungen berechnet wurden (links) sowie benötigte Molekül-Struktureigenschaften
© Fraunhofer IWM
Um einen Schmierstoff neu zu entwickeln oder zu verbessern ist es wichtig, sein Verhalten genau zu kennen: Druck, Temperatur, Scherrate und Viskosität sind dabei wichtige Größen. »Den Schmierfilm experimentell zu vermessen ist sehr schwer, weil die dazu benötigen Bedingungen kaum kontrolliert einzustellen sind«, erklärt Dr. Kerstin Falk, Mitarbeiterin der Gruppe Multiskalenmodellierung und Tribosimulation des Freiburger Fraunhofer-Instituts für Werkstoffmechanik IWM. »In einigen Bereichen treten zum Beispiel so extreme Pressungen auf, dass lokal ein Druck im Giga-Pascal-Bereich entsteht«, so die Expertin für Tribologie, der Lehre von Reibung und Verschleiß. Simulationen können dort weiterhelfen, wo Experimente schwierig sind: Bisher wurden Vorhersagen für das Schmierstoffverhalten bei hohem Druck meist aus »Hochrechnungen« der Ergebnisse von Experimenten in normalem Druckbereich gewonnen. Leider versagt dieses Vorgehen jedoch bei den extrem hohen Drücken, die lokal in geschmierten Reibkontakten auftreten.
Viskosität bei hohem Druck exakt berechnen
Mit atomistischen Molekulardynamiksimulationen kann jedoch die Physikerin Schmierstoffeigenschaften wie die Viskosität unter kontrollierten, beliebig einstellbaren Druck- und Temperaturbedingungen vorhersagen. »Wir haben die Viskosität sehr erfolgreich für verschiedene Modellschmierstoffe bei Druck bis hin zu 700 Mega-Pascal berechnet«, sagt Dr. Kerstin Falk. »Unsere Modell-Schmierstoffe bestanden aus einfachen linearen oder aus verzweigten Alkanen«, erklärt sie weiter. Alkane oder Paraffine sind sehr stabile Ketten aus Kohlenstoff und Wasserstoff, und bilden den Grundbestandteil vieler gängiger Öle und Kraftstoffe.
Schmierstoffe charakterisieren aufgrund ihrer Moleküleigenschaften
Doch Falks eigentliches Ziel war, ein praktikableres Vorhersagemodell als bisher zu erhalten. Es sollte die Berechnung der Viskositätswerte für bestimmte Bedingungen im Reibspalt erlauben, ohne erst aufwändige Molekulardynamiksimulationen durchführen zu müssen. Dazu nutzten Dr. Kerstin Falk und ihre Kollegen Dr. Daniele Savio und Prof. Michael Moseler atomistische Simulationen, die quasi wie eine »virtuelle Superlupe« Einblicke auf die mikroskopische Struktur und Dynamik der Schmierstoffmoleküle ermöglichen. So fand das Team heraus, welche drei molekularen Eigenschaften eines Schmierstoffs seine Viskosität maßgeblich bestimmen: die Molekül-Querschnittsflächen, die Kettenflexibilität und der Abstand zwischen den einzelnen Molekülen. Sind diese Eigenschaften einmal bestimmt, lässt sich damit die Viskosität eines Schmierstoffs bei verschiedensten Bedingungen einfach und genau berechnen. »Umgekehrt benutzt können wir mit dieser Simulationsmethode auch die passenden Moleküle für bestimmte tribologische Beanspruchungen finden«, sagt Dr. Kerstin Falk. »Und in Kombination mit zusätzlichen quantenchemischen Simulationen zur Wechselwirkung des Schmierstoffs mit Bauteiloberflächen können wir unseren Kundinnen und Kunden den passenden Schmierstoff für ihre Anwendung vorschlagen«, fügt Prof. Michael Moseler hinzu.
In Zukunft soll mit diesen neuen Ergebnissen als Basis auch das Schmierstoffverhalten bei sehr großen Geschwindigkeiten und damit großen Scherraten untersucht werden. Und es wird betrachtet, wie sich ein Schmierstoff in einem sehr engen Reibspalt verhält, der nur wenige Moleküldurchmesser breit ist. Auch andere Schmierstoffe können Dr. Kerstin Falk und ihre Kollegen mit der neuen Methode charakterisieren – beispielsweise wasserbasierte Schmierstoffe für umweltverträgliche Anwendungen.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft





























































