Neue Methode zur hochgenauen Vorhersage von NMR-Parametern
Hochgenaue Ergebnisse bei niedrigem Rechenaufwand
Die Kernresonanzspektroskopie (“Nuclear Magnetic Resoncance” – NMR) ist eine der wichtigsten analytischen Techniken in der Chemie, die in der pharmazeutischen Industrie, der Biomedizin oder den Materialwissenschaften nicht fehlen darf. Sie dient dazu, die Struktur und Dynamik von Molekülen aufzuklären. NMR-Spektren allein genügen jedoch oft nicht für eine vollständige Strukturbestimmung und so müssen Forscher quantenchemische Berechnungen zur Ergänzung und Interpretation nutzen. Die theoretischen Chemiker simulieren dabei als wichtigsten Parameter die „chemische Verschiebung“ für jedes Atom, um genaueste Informationen zu erlangen. Doch die bisher genutzten Methoden gelangten immer wieder an Grenzen.
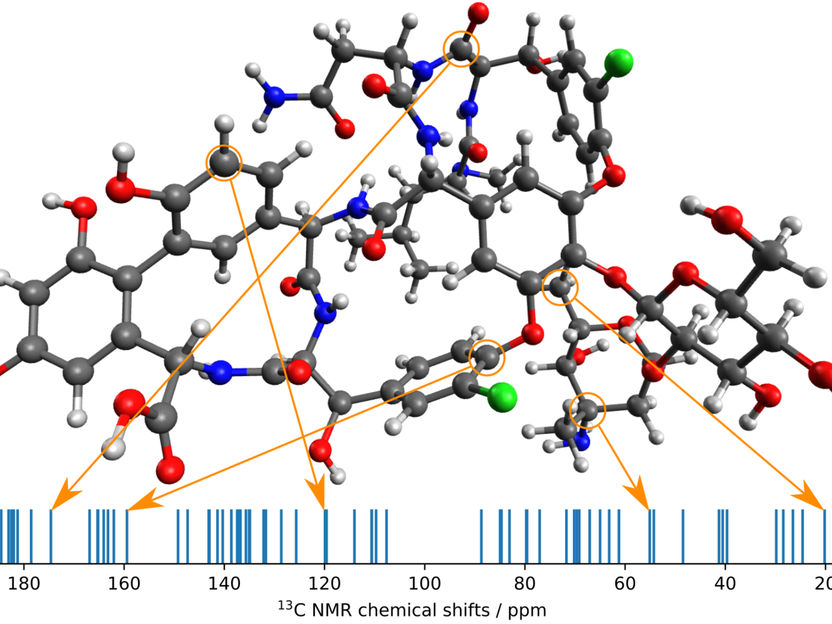
Die neue Methode der MPI Forscher in der Anwendung. Ein Konformer des Vancomycin-Moleküls und sein berechnetes 13C-NMR-Spektrum, bei dem einige Werte den Atomen zugeordnet sind.
© MPI für Kohlenforschung
Bisherige Ergebnisse oft zu ungenau oder nur mit hohem Rechenaufwand erzielbar - die neue DLPNO-Näherung reduziert den Rechenaufwand genauer Methoden erheblich
Die Kombination aus Experiment und immer neuen theoretischen Methoden führt zu Erkenntnissen, die auf anderem Wege nicht zugänglich gewesen wären. Allerdings stehen Wissenschaftler häufig vor einem Dilemma: Effiziente Methoden wie die Dichtefunktionaltheorie – das Arbeitspferd der Theoretischen Chemie – liefern häufig nur unzureichende Genauigkeit. Hochgenaue Wellenfunktionsmethoden wiederum nehmen enorme Rechenressourcen in Anspruch. Zudem können sie nicht für große Moleküle wie z. B. in der Pharmazeutik oder Biomedizin angewendet werden.
Forscher der Abteilung für Molekulare Theorie und Spektroskopie am MPI für Kohlenforschung entwickelten nun einen neuen Ansatz, der den Durchbruch bringt. Ihre Wellenfunktionsmethode zur Vorhersage von NMR- Parametern basiert auf dem von Frank Neese beschriebenen Konzept der Domänen-basierten Lokalen Paar Natürlichen Orbitalen (DLPNO) Näherung. Die Theoretiker um Stoychev et al. erreichten hochgenaue Ergebnisse bei niedrigem Rechenaufwand. In einem kürzlich veröffentlichten Artikel beschreiben sie, wie ihnen die exakte Berechnung chemischer Verschiebungen für Systeme mit hunderten Atomen gelang. Die neue Methode eröffnet interessante Perspektiven für Anwender, denn in vielen Bereichen der Chemie gehört die Interpretation von NMR-Spektren zum Tagesgeschäft.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News










Weitere News von unseren anderen Portalen






























Verwandte Inhalte finden Sie in den Themenwelten
Themenwelt Spektroskopie
Durch die Untersuchung mit Spektroskopie ermöglicht uns einzigartige Einblicke in die Zusammensetzung und Struktur von Materialien. Von der UV-Vis-Spektroskopie über die Infrarot- und Raman-Spektroskopie bis hin zur Fluoreszenz- und Atomabsorptionsspektroskopie - die Spektroskopie bietet uns ein breites Spektrum an analytischen Techniken, um Substanzen präzise zu charakterisieren. Tauchen Sie ein in die faszinierende Welt der Spektroskopie!

Themenwelt Spektroskopie
Durch die Untersuchung mit Spektroskopie ermöglicht uns einzigartige Einblicke in die Zusammensetzung und Struktur von Materialien. Von der UV-Vis-Spektroskopie über die Infrarot- und Raman-Spektroskopie bis hin zur Fluoreszenz- und Atomabsorptionsspektroskopie - die Spektroskopie bietet uns ein breites Spektrum an analytischen Techniken, um Substanzen präzise zu charakterisieren. Tauchen Sie ein in die faszinierende Welt der Spektroskopie!