Programm für maschinelles Lernen in Spielen inspiriert zur Entwicklung eines bahnbrechenden wissenschaftlichen Tools
Ein neues KI-Tool modelliert in Rekordzeit das Verhalten von Clustern aus Nanopartikeln
Wir lernen neue Fähigkeiten durch Wiederholung und Verstärkung. Durch Versuch und Irrtum wiederholen wir Handlungen, die zu guten Ergebnissen führen, versuchen schlechte Ergebnisse zu vermeiden und versuchen, die dazwischen liegenden zu verbessern. Forscher entwickeln jetzt Algorithmen, die auf einer Form von künstlicher Intelligenz beruhen, die das Verstärkungslernen nutzt. Sie wenden sie an, um die chemische Synthese und die Entdeckung von Medikamenten zu automatisieren und sogar Spiele wie Schach und Go zu spielen.
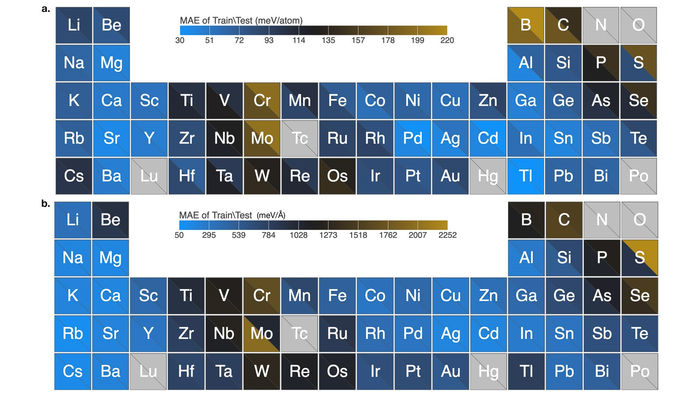
Das Diagramm zeigt die hervorragende Leistung des Algorithmus für Kraftfeldvorhersagen von elementaren Nanoclustern, die 54 Elemente des Periodensystems abdecken. Die Daten zeigen einen sehr niedrigen mittleren absoluten Fehler.
Image by Argonne National Laboratory
Wissenschaftler am Argonne National Laboratory des US-Energieministeriums (DOE) haben einen Algorithmus für verstärkendes Lernen für eine weitere Anwendung entwickelt. Er dient der Modellierung der Eigenschaften von Materialien auf atomarer und molekularer Ebene und dürfte die Materialforschung erheblich beschleunigen.
Wie der Mensch "lernt" dieser Algorithmus das Lösen von Problemen aus seinen Fehlern und Erfolgen. Aber er tut dies ohne menschliches Zutun.

In der Vergangenheit war Argonne weltweit führend im Bereich der molekularen Modellierung. Dazu gehört die Berechnung der Kräfte zwischen den Atomen in einem Material und die Verwendung dieser Daten zur Simulation seines Verhaltens unter verschiedenen Bedingungen im Laufe der Zeit.
In der Vergangenheit stützten sich solche Modelle jedoch stark auf menschliche Intuition und Fachwissen und erforderten oft jahrelange mühsame Arbeit. Der Algorithmus des Teams für verstärkendes Lernen reduziert den Zeitaufwand auf Tage und Stunden. Außerdem liefert er hochwertigere Daten als herkömmliche Methoden.
"Wir haben uns von AlphaGo inspirieren lassen", sagt Sukriti Manna, Forschungsassistentin im Center for Nanoscale Materials (CNM) von Argonne, einer Nutzereinrichtung des DOE Office of Science. "Es ist das erste Computerprogramm, das einen Go-Weltmeister besiegt hat."
Das Standard-Go-Brett hat 361 Positionsfelder, viel mehr als die 64 Felder eines Schachbretts. Daraus ergibt sich eine große Anzahl möglicher Brettkonfigurationen. Der Schlüssel dazu, dass AlphaGo Weltmeister wurde, war seine Fähigkeit, seine Fähigkeiten durch verstärkendes Lernen zu verbessern.
Die Automatisierung der molekularen Modellierung ist natürlich etwas ganz anderes als ein Go-Computerprogramm. Eine der Herausforderungen, mit denen wir konfrontiert waren, ähnelt der Entwicklung des für selbstfahrende Autos erforderlichen Algorithmus", so Subramanian Sankaranarayanan, Gruppenleiter am CNM in Argonne und außerordentlicher Professor an der University of Illinois Chicago.
Während das Go-Board statisch ist, ändert sich die Verkehrsumgebung ständig. Das selbstfahrende Auto muss mit anderen Autos, unterschiedlichen Routen, Verkehrsschildern, Fußgängern, Kreuzungen und so weiter interagieren. Die Parameter für die Entscheidungsfindung ändern sich mit der Zeit ständig.
Auch bei der Lösung schwieriger realer Probleme im Bereich der Materialentdeckung und des Materialdesigns müssen ständig Entscheidungen getroffen werden, um optimale Lösungen zu finden. In den Algorithmus des Teams sind Entscheidungsbäume integriert, die je nach Erfolg bei der Optimierung der Modellparameter eine positive Verstärkung ausschütten. Das Ergebnis ist ein Modell, das die Materialeigenschaften und ihre Veränderungen im Laufe der Zeit genau berechnen kann.
Das Team testete seinen Algorithmus erfolgreich mit 54 Elementen des Periodensystems. Ihr Algorithmus lernte, wie man Kraftfelder von Tausenden von nanoskaligen Clustern für jedes Element berechnet und führte die Berechnungen in Rekordzeit durch. Diese Nanocluster sind bekannt für ihre komplexe Chemie und die Schwierigkeiten, die herkömmliche Methoden bei ihrer genauen Modellierung haben.
"Das ist so, als würde man die Berechnungen für mehrere Doktorarbeiten in wenigen Tagen statt in Jahren durchführen", so Rohit Batra, CNM-Experte für datengesteuerte und maschinelle Lernverfahren. Das Team hat diese Berechnungen nicht nur für Nanocluster aus einem einzigen Element, sondern auch für Legierungen aus zwei Elementen durchgeführt.
"Unsere Arbeit stellt einen großen Schritt nach vorn bei der Entwicklung solcher Modelle für die Materialwissenschaft dar", so Sankaranarayanan. "Die Qualität unserer Berechnungen für die 54 Elemente mit dem Algorithmus ist viel höher als der Stand der Technik."
Die Ausführung des Algorithmus des Teams erforderte Berechnungen mit großen Datensätzen auf Hochleistungsrechnern. Zu diesem Zweck griff das Team auf den Carbon-Cluster des CNM und den Theta-Supercomputer in der Argonne Leadership Computing Facility, einer Einrichtung des DOE Office of Science, zurück. Außerdem griffen sie auf die Rechenressourcen des National Energy Research Scientific Computing Center zurück, einer Einrichtung des DOE Office of Science am Lawrence Berkeley National Laboratory.
"Der Algorithmus dürfte die Zeit, die für die Bewältigung großer Herausforderungen in vielen Bereichen der Materialwissenschaft benötigt wird, erheblich verkürzen", sagte Troy Loeffler, ein Chemiker für Berechnungen und theoretische Chemie im CNM. Beispiele sind Materialien für elektronische Geräte, Katalysatoren für industrielle Prozesse und Batteriekomponenten.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

NANOPHOX CS von Sympatec
Partikelgrößenanalyse im Nanobereich: Hohe Konzentrationen problemlos analysieren
Zuverlässige Ergebnisse ohne aufwändige Probenvorbereitung

DynaPro Plate Reader III von Wyatt Technology
Screening von Biopharmazeutika und anderen Proteinen mit automatisierter dynamischer Lichtstreuung
Hochdurchsatz-DLS/SLS-Messungen von Lead Discovery bis Qualitätskontrolle

Eclipse von Wyatt Technology
FFF-MALS System zur Trennung und Charakterisierung von Makromolekülen und Nanopartikeln
Neuestes FFF-MALS-System entwickelt für höchste Benutzerfreundlichkeit, Robustheit und Datenqualität









































