Künstliche Intelligenz hilft, die Grenzen der Chemie zu erforschen
Maschinelles Lernen hilft bei der Simulation reaktiver Molekulardynamik für Forschung und Entdeckung
Die Möglichkeit, das Verhalten von Systemen auf atomarer Ebene zu simulieren, ist ein leistungsfähiges Instrument für alle Bereiche von der Entwicklung von Arzneimitteln bis hin zur Materialforschung. Ein Team unter der Leitung von Forschern des Los Alamos National Laboratory hat maschinell lernende interatomare Potenziale entwickelt, die molekulare Energien und auf Atome wirkende Kräfte vorhersagen und so Simulationen ermöglichen, die im Vergleich zu bestehenden Berechnungsmethoden Zeit und Kosten sparen.
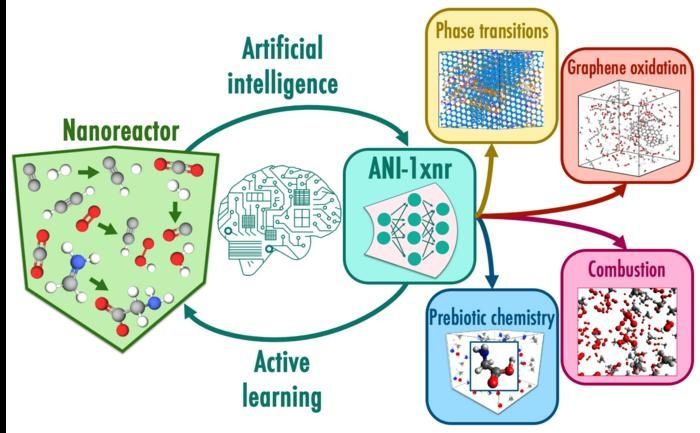
In diesem Arbeitsablauf werden bei Nanoreaktorsimulationen automatisch reaktive chemische Räume abgetastet, ohne dass man sich auf menschliche Intuition verlassen muss. Der Nanoreaktor ist eine spezielle Klasse von atomistischen Simulationen, in denen chemische Reaktionen durch kollidierende Moleküle bei hohen Geschwindigkeiten ausgelöst werden. Aktives Lernen nutzt das Potenzial des maschinellen Lernens, ANI-1xnr, um die Dynamik des Nanoreaktors zu steuern und Strukturen mit hohen Unsicherheiten auszuwählen. Fallstudien wie Phasenübergänge bei der Verbrennung von Kohlenstoff und Methan testen die Allgemeingültigkeit des resultierenden Modells ANI-1xnr.
Los Alamos National Laboratory
"Maschinelle Lernpotenziale bieten zunehmend eine effektive Alternative zu rechenintensiven Simulationen, die komplexe physikalische Systeme auf atomarer Ebene abzubilden versuchen", so Benjamin Nebgen, Chemiephysiker bei Los Alamos und Mitautor eines kürzlich erschienenen Nature Chemistry-Artikels, der die Arbeit beschreibt. "Ein allgemeines reaktives interatomares Lernpotenzial, das auf ein breites Spektrum der reaktiven Chemie anwendbar ist, ohne dass eine Neuanpassung erforderlich ist, wird der Chemie und der Materialwissenschaft großen Nutzen bringen."
Überbrückung der Lücke bei effektiven Simulationen
Die Erstellung effektiver Simulationen für die Molekulardynamik in der Chemie erfolgt traditionell mit physikalisch basierten Berechnungsmodellen, einschließlich klassischer Kraftfelder oder der Quantenmechanik. Quantenmechanische Modelle sind zwar genau und allgemein anwendbar, aber extrem rechenintensiv. Im Gegensatz dazu sind klassische Kraftfelder zwar rechnerisch effizient, aber relativ ungenau und nur auf eine begrenzte Anzahl von Systemen anwendbar. ANI-1xnr, das transformative maschinelle Lernmodell des Teams, schließt die Lücke in Bezug auf Geschwindigkeit, Genauigkeit und Allgemeinheit, die in der Chemie seit vielen Jahrzehnten besteht. (Maschinelles Lernen ist eine Anwendung der künstlichen Intelligenz, bei der Computerprogramme durch Training "lernen".)
ANI-1xnr ist das erste reaktive, maschinell lernende interatomare Potenzial, das allgemein genug ist - es kann auf viele verschiedene chemische Systeme angewandt werden -, um mit physikbasierten Berechnungsmodellen für die Durchführung groß angelegter reaktiver atomistischer Simulationen zu konkurrieren. ANI-1xnr wurde mit Hilfe eines automatisierten Arbeitsablaufs entwickelt, der reaktive Molekulardynamiksimulationen für ein breites Spektrum chemischer Systeme mit den Elementen Kohlenstoff, Wasserstoff, Stickstoff und Sauerstoff durchführte.
ANI-1xnr erwies sich als fähig, ein breites Spektrum von Systemen zu untersuchen, von Kohlenstoff-Phasenübergängen über Verbrennung bis hin zu präbiotischer Chemie. Das Team validierte die Simulationen, indem es sie mit Ergebnissen aus Experimenten und konventionellen Berechnungsmethoden verglich.
Ein interatomares Transformationspotenzial
"ANI-1xnr erfordert keine Fachkenntnisse oder Anpassungen für jeden neuen Anwendungsfall und ermöglicht es Wissenschaftlern aus den verschiedensten Bereichen, unbekannte chemische Prozesse zu untersuchen", so Richard Messerly, Computerwissenschaftler in Los Alamos und Mitverfasser der Studie. "Die allgemeine Anwendbarkeit von ANI-1xnr ist revolutionär und stellt einen bedeutenden Schritt auf dem Weg zur Ablösung der langjährigen Modellierungstechniken für die Untersuchung reaktiver Chemie in großem Maßstab dar."
Der von dem Team verwendete Datensatz und der ANI-1xnr-Code wurden der Forschungsgemeinschaft öffentlich zugänglich gemacht.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung
Shuhao Zhang, Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Olexandr Isayev, Nicholas Lubbers, Richard A. Messerly, Justin S. Smith; "Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential"; Nature Chemistry, 2024-3-7
Weitere News aus dem Ressort Forschung & Entwicklung






























































