Chemisches Chamäleon gebändigt
Computersimulationen zeigen: Protoniertes Methan wird durch Mikrosolvatation gezähmt
Wie man das Chamäleon unter den Molekülen dazu bekommt, sich auf ein bestimmtes „Aussehen“ festzulegen, haben RUB-Chemiker um Professor Dominik Marx herausgefunden. Das Molekül CH5+ ist normalerweise nicht durch eine einzige starre Struktur zu beschreiben, sondern dynamisch flexibel. Mit Computersimulationen zeigte das Team vom Lehrstuhl für Theoretische Chemie, dass CH5+ eine bestimmte Struktur annimmt, sobald man Wasserstoffmoleküle anlagert. „Damit haben wir einen wichtigen Schritt getan, um in Zukunft experimentelle Schwingungsspektren zu verstehen“, sagt Dominik Marx.
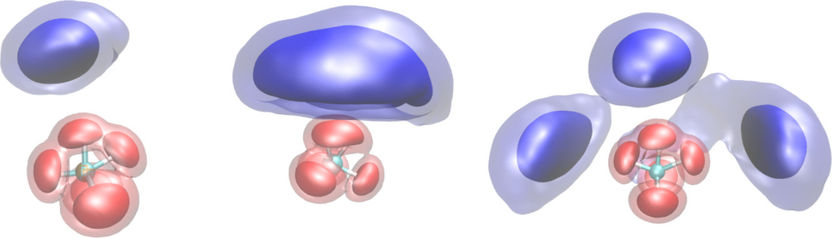
Je nachdem, wie viele H2-Lösungsmittelmoleküle (blau) sich an das CH5+-Molekül anlagern, verändert sich der Bereich, in dem sich die Wasserstoffe des CH5+-Moleküls bewegen (rot); seine Struktur wird also teilweise „eingefroren“. Die Flächen repräsentieren quantenmechanische Aufenthaltswahrscheinlichkeitsdichten bei einer Temperatur von 20 Kelvin.
© A. Witt, S. Ivanov, D. Marx
Im CH5+-Molekül sind die Wasserstoffatome permanent auf Wanderschaft
Die Supersäure CH5+, auch protoniertes Methan genannt, kommt im Weltall vor – dort, wo sich neue Sterne bilden. Forscher entdeckten das Molekül schon in den 1950er-Jahren, doch viele seiner Eigenschaften sind nach wie vor unbekannt. Anders als herkömmliche Moleküle, in denen alle Atome eine feste Position besitzen, bewegen sich die fünf Wasserstoffatome in CH5+ ständig um das Kohlenstoffzentrum. Wissenschaftler sprechen von „hydrogen scrambling“. Diese dynamisch flexible Struktur klärten die Arbeitsgruppen von Dominik Marx und Stefan Schlemmer von der Universität Köln im Rahmen einer langjährigen Zusammenarbeit auf. Nun wollte Marx‘ Team wissen, ob sich die Struktur unter bestimmten Bedingungen durch Anlagerung von Lösungsmittelmolekülen „einfrieren“ lässt – ein Prozess, der Mikrosolvatation heißt.
Mikrosolvatation: Anlagerung von Wasserstoffmolekülen an CH5+
Zu diesem Zweck umgaben die Chemiker das CH5+-Molekül virtuell mit einigen wenigen Wasserstoffmolekülen (H2). Dabei passiert im Ergebnis das Gleiche, wie wenn man normale Ionen in Wasser löst: An jedes Ion lagert sich eine relativ fest gebundene Hülle aus Wassermolekülen an, um anschließend einzelne Ionen mit einigen daran gebundenen Lösungsmittelmolekülen in die Gasphase zu transferieren. Um die CH5+-Wasserstoff-Komplexe zu beschreiben, reichen klassische ab initio-Molekulardynamik-Simulationen nicht aus. Denn das „hydrogen scrambling“ beruht auf Quanteneffekten. Daher nutzte Marx‘ Gruppe eine selbst entwickelte, voll quantenmechanische Methode, die sogenannten ab initio-Pfadintegralsimulationen. Mit ihr lassen sich die essenziellen Quanteneffekte abhängig von der Temperatur in die Rechnung einbeziehen.
Wasserstoffmoleküle verleihen dem CH5+-Molekül Struktur
Die Chemiker führten die Simulationen für eine Temperatur von 20 Kelvin durch; das entspricht -253 Grad Celsius. In nicht gelöster Form tauschen die Wasserstoffatome im CH5+-Molekül auch bei so niedrigen Temperaturen permanent ihre Positionen – und zwar ausschließlich aufgrund quantenmechanischer Effekte. Wenn CH5+ von Wasserstoff umgeben ist, wird das „hydrogen scrambling“ jedoch stark beeinflusst und kann sogar ganz zum Erliegen kommen: Das Molekül nimmt eine rudimentäre Struktur an. Wie genau diese aussieht, hängt davon ab, wie viele Wasserstoffmoleküle sich an ein CH5+-Molekül anlagern. „Mich interessiert nun besonders, ob superflüssiges Helium – ähnlich wie hier der Wasserstoff – auch die Wanderung der Wasserstoffe im CH5+ stoppen kann“, sagt Marx. Experimentell arbeitende Forscher nutzen superflüssiges Helium, um hochaufgelöste Spektren von darin eingelagerten Molekülen zu messen. Für CH5+ ist das bislang aber nicht möglich ist. In der superflüssigen Phase sind die Heliumatome allerdings aufgrund quantenstatistischer Effekte nicht unterscheidbar. Um diese Tatsache beschreiben zu können, entwickelten die Theoretischen Chemiker an der RUB über viele Jahre hinweg eine neue, noch aufwändigere pfadintegralbasierte Simulationsmethode, die seit kurzem auch auf reale Fragestellungen angewendet wird.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft





















































