Steuerbare Katalyse: Die Lösung des Teilchengrößen-Problems
An der TU Wien gelang es, mikroskopische und makroskopische Betrachtungsweisen zu verbinden – und dadurch ein altes Rätsel zu lösen
chemische Reaktionen kann man auf unterschiedlichen Ebenen betrachten: Auf der Ebene einzelner Atome und Moleküle zeigt sich, welche neuen Verbindungen möglich sind. Auf der Ebene von winzigen Teilchen auf Nano- und Mikrometer-Skala kann man verstehen, wie Katalysator-Materialien in die chemische Reaktion eingreifen. Und wenn man die chemische Reaktion großtechnisch nutzen will, braucht man den Blick auf die makroskopische Skala.
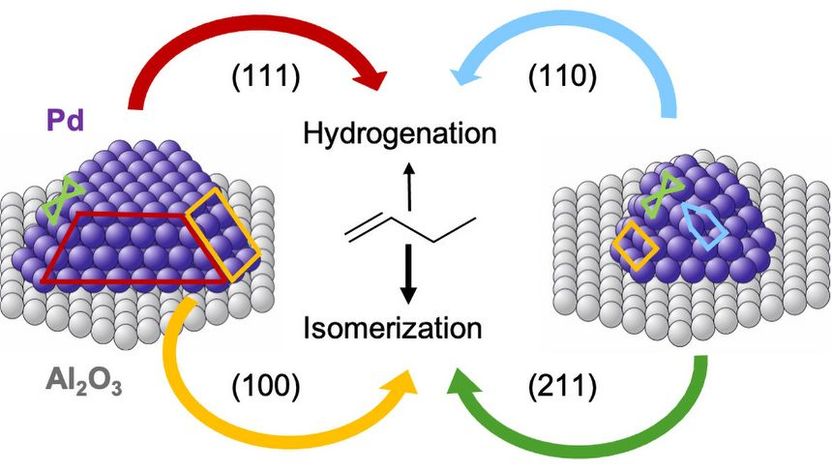
Palladium Nanokristalle auf Aluminiumoxid: Manche Facetten ermöglichen die Isomerisierung von 1-Buten zu 2-Buten, andere hingegen die Hydrierung zu Butan.
Technische Universität Wien
Normalerweise verwendet man für jeden Bereich unterschiedliche Zugänge getrennt voneinander. Doch für komplizierte chemische Reaktionen auf Katalysator-Oberflächen genügt das nicht. An der TU Wien gelang nun ein wichtiger Schritt: Erstmals konnten alle Ebenen von der Mikro- bis zur Makroebene miteinander verbunden werden, um eine technisch wichtige chemische Reaktion bei realen Bedingungen vollständig zu beschreiben. Dadurch konnte man auch erklären, warum die Größe von Katalysator-Partikeln eine entscheidende Rolle spielt. Die Ergebnisse wurden nun im Fachjournal „Nature Communications“ veröffentlicht.
Isomere: Gleiche Atome, unterschiedliche Moleküle
Von vielen Molekülen gibt es unterschiedliche Varianten: Dieselben Atome lassen sich auf unterschiedliche Weise anordnen, man spricht dann von „Isomeren“. Es ist wichtig, zwischen diesen Isomeren zu unterscheiden – so ist etwa ein bestimmtes Isomer des Kohlenwasserstoffs Buten günstig für die Treibstoffherstellung, eine andere Buten-Variante möchte man hingegen haben, wenn man Polymere produzieren will. Genau die richtigen Isomere zu erzeugen oder ein Isomer in ein anderes umzuwandeln ist eine heikle Aufgabe, die mit ganz bestimmten Katalysatoren gelingen kann.
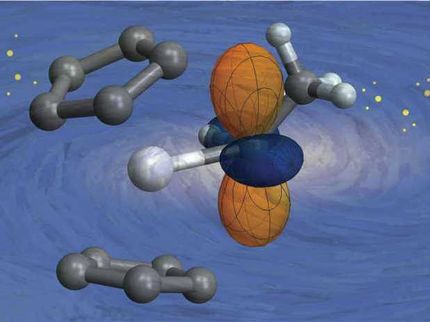
„Ein besonders wichtiger Katalysator für solche Prozesse ist Palladium“, sagt Prof. Günther Rupprechter vom Institut für Materialchemie der TU Wien. „Normalerweise trägt man es in Form winziger Nanokristalle auf einen Träger auf. Bestimmte Moleküle werden dann von diesen Körnchen festgehalten, und das ermöglicht die chemische Reaktion“.
Bekannt ist, dass oft eine bestimmt Partikelgröße für die katalytische Wirkung ausschlaggebend ist, doch eine detaillierte Erklärung dafür, wie das funktioniert, gab es bisher nicht. „Die technisch genutzten Teilchen bestehen einfach aus zu vielen Atomen, um sie quantenchemisch am Computer berechnen zu können“, erklärt Dr. Alexander Genest, der Erstautor der aktuellen Studie. „Man muss daher neue Wege finden, um die unterschiedlichen Betrachtungsweisen miteinander zu kombinieren“.
Realistische Bedingungen statt idealisierter Systeme
Das Forschungsteam der TU Wien und die Kooperationspartner aus Singapur, Alicante und München wählten für die Untersuchungen eine heikle aber wichtige Reaktion aus: Die Isomerisierung von Alkenen. „Das ist besonders herausfordernd, weil es verschiedene Reaktionspfade gibt, die gleichzeitig eine Rolle spielen“, sagt Günther Rupprechter. „Wichtig war uns auch, die Reaktion bei realistischen Bedingungen zu studieren: In bisherigen Grundlagen-Untersuchungen analysierte man Reaktionen oft im (Ultra-)Hochvakuum, bei tiefen Temperaturen. Doch in der Praxis hat man es mit ganz anderen Parametern zu tun. Wir wollten daher wissen, wie diese Isomerisierung bei Atmosphärendruck und 100 °C abläuft“.
Man beginnt auf Ebene der Atome und Moleküle: „Mit Hilfe der Dichtefunktionaltheorie können wir die elementaren Reaktionsschritte der Moleküle berechnen, die sich an unterschiedlichen Facetten der Palladium-Kristalle anlagern“, sagt Alexander Genest. Diese Berechnungen fließen dann in sogenannte mikrokinetische Modelle ein, mit denen man die Dynamik chemischer Reaktionen auf einer größeren Zeitskala im Computer ablaufen lassen kann. Und aus diesen Ergebnissen wiederum lässt sich dann auf die Gesamtmenge der gewünschten chemischen Produkte schließen, die nach einer bestimmten Zeit bei bestimmten Parametern vorhanden sein wird.
„Die Modellrechnungen stimmen nicht nur qualitativ, sondern auch quantitativ sehr gut mit unseren experimentellen Messungen überein“, betont Prof. Günther Rupprechter. „Das ist ein wichtiger Durchbruch – bisher war das so nicht möglich“. Nun kann man im Detail erklären, warum unterschiedliche Größen der Palladium-Partikel sich unterschiedlich auf die chemischen Abläufe auswirken: Große Körnchen haben glatte Oberflächen, während kleinere Körnchen eher rundlich und gestuft sind. Die Anordnung der Palladium-Atome in unterschiedlichen Geometrien beeinflusst die Reaktionsenergie und somit das katalytische Verhalten.
Optimale Resultate statt Versuch und Irrtum
„Wenn man in der Industrie einen chemischen Prozess optimiert, ist man oft auf Versuch und Irrtum angewiesen“, sagt Günther Rupprechter. „Bei welchen Parametern lässt man die Reaktion ablaufen? Welche Katalysatoren verwendet man – und in welcher Form? Das sind Fragen, die man bisher auf theoretischer Ebene kaum beantworten konnte“. Man probiert also verschiedene Varianten aus und wählt dann die erfolgreichste. Wenn man dann aber einen Prozess vom Labormaßstab auf industriellen Maßstab hochskalieren möchte, ändert sich vieles, und man weiß wieder nicht, welche Bedingungen die besten sind.
„Wir haben nun gezeigt, dass man solche Abläufe umfassend verstehen kann, wenn man verschiedene Betrachtungsebenen miteinander verknüpft“, sagt Alexander Genest. „Diese Herangehensweise ist natürlich auch auf viele andere katalytische Reaktionen anwendbar“. In der chemischen Industrie soll es somit möglich werden, durch Modellierung am Computer chemische Herstellungsverfahren zu optimieren und gleichzeitig teure und zeitraubende Versuchsreihen auf ein Minimum zu reduzieren.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft