Neuartiges maschinelles Lernmodell für die Molekularsimulation
Das Forschungsteam von Prof. JIANG Bin an der University of Science and Technology of China (USTC) hat ein universelles feldinduziertes rekursiv eingebettetes neuronales Netzwerkmodell (FIREANN) entwickelt, das die Wechselwirkungen zwischen System und Feld mit hoher Effizienz simulieren kann.
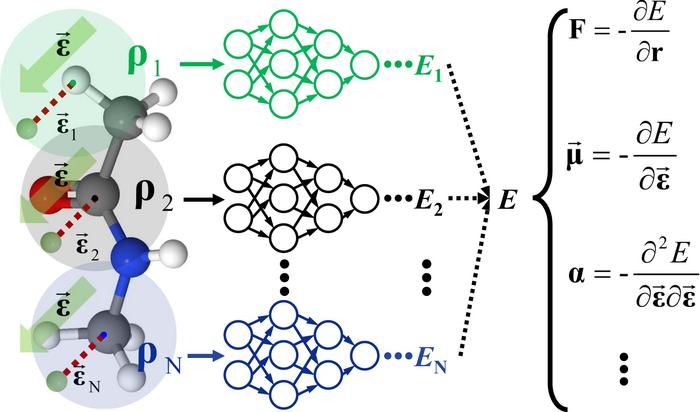
Schematische Darstellung des FIREANN-Rahmens.
Prof. JIANG’s team
Atomsimulationen spielen eine entscheidende Rolle beim Verständnis der Spektren und der Dynamik komplexer chemischer, biologischer und materieller Systeme auf mikroskopischer Ebene. Der Schlüssel zu atomaren Simulationen ist die genaue Darstellung hochdimensionaler potentieller Energieoberflächen (PES). In den letzten Jahren hat sich die Verwendung atomistischer Modelle des maschinellen Lernens (ML) zur genauen Darstellung von PESs durchgesetzt. Die meisten ML-Modelle beschreiben jedoch nur isolierte Systeme und können die Wechselwirkungen zwischen externen Feldern und den Systemen nicht erfassen, die die chemische Struktur verändern und den Phasenübergang durch feldinduzierte elektronische oder Spin-Polarisation steuern können. Ein neuartiges ML-Modell, das externe Felder berücksichtigt, ist dringend erforderlich.
Um dieses Problem zu lösen, schlug das Forschungsteam von Prof. JIANG einen "All-in-one"-Ansatz vor. Das Team behandelte zunächst externe Felder als virtuelle Atome und verwendete eingebettete Atomdichten (EADs) als Deskriptoren für die atomare Umgebung. Die feldinduzierte EAD (FI-EAD) wurde aus der linearen Kombination der feldabhängigen Orbitale und der koordinatenbasierten Orbitale der Atome abgeleitet, die die Art der Wechselwirkung zwischen dem externen Feld und dem System erfasst, was zur Entwicklung des FIREANN-Modells führte.
Dieses Modell korreliert verschiedene Reaktionseigenschaften des Systems, wie Dipolmoment und Polarisierbarkeit, genau mit den potenziellen Energieänderungen, die von externen Feldern abhängen, und bietet so ein genaues und effizientes Werkzeug für die Spektroskopie und die Simulation der Dynamik komplexer Systeme unter externen Feldern.
Das Team überprüfte die Fähigkeit des FIREANN-Modells durch dynamische Simulationen von N-Methylacetamid und flüssigem Wasser unter starken externen elektrischen Feldern, die beide eine hohe Genauigkeit und Effizienz zeigten. Es ist erwähnenswert, dass das FIREANN-Modell bei periodischen Systemen das inhärente Problem der Mehrwertigkeit der Polarisation überwinden kann, indem es nur mit den Daten der atomaren Kräfte trainiert.
Diese Forschung füllte die Lücke der fehlenden genauen Darstellung des externen Feldes in ML-Modellen, was zum Fortschritt der molekularen Simulationen in der Chemie, Biologie und Materialwissenschaft beitragen wird.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft






























































